Modeling the Electron Transfer Chain in an Artificial Photosynthetic Machine
The Journal of Physical Chemistry Letters
DOI: 10.1021/acs.jpclett.0c02766
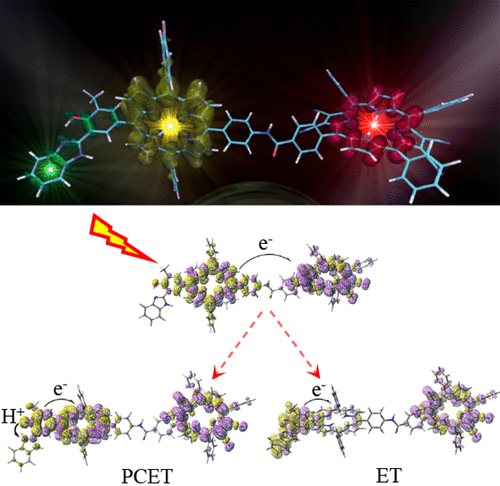
The development of efficient artificial leaves relies on the subtle combination of molecular assemblies able to absorb sunlight, converting light energy into electrochemical potential energy and finally transducing it into accessible chemical energy. The electronic design of these charge transfer molecular machines is crucial to build a complex supramolecular architecture for the light energy conversion. Here, we present an ab initio simulation of the whole decay pathways of a recently proposed artificial molecular reaction center. A complete structural and energetic characterization has been carried out with methods based on density functional theory, its time-dependent version, and a broken-symmetry approach. On the basis of our findings we provide a revision of the pathway only indirectly postulated from an experimental point of view, along with unprecedented and significant insights on the electronic and nuclear structure of intramolecular charge-separated states, which are fundamental for the application of this molecular assembly in photoelectrochemical cells. Importantly, we unravel the molecular driving forces of the various charge transfer steps, in particular those leading to the proton-coupled electron transfer final product, highlighting key elements for the future design strategies of such molecular assays.