Comparing the performance of TD-DFT and SAC-CI methods in the description of excited states potentia
Journal of Computational Chemistry 2017, 38 (14), 1084-1092
DOI: 10.1002/jcc.24780
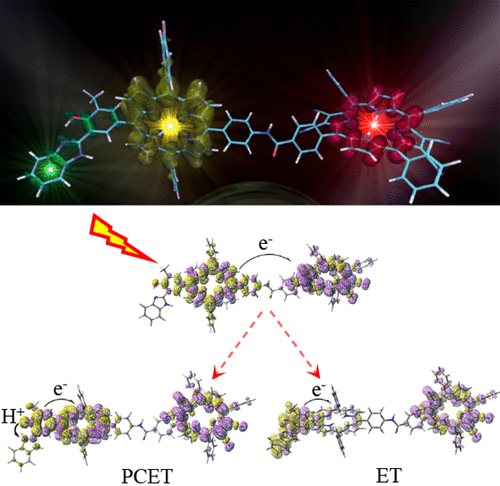
The performances, in the description of excited state potential energy surfaces, of several density functional approximations representative of the currently most applied exchange correlation functionals’ families have been tested with respect to post Hartree-Fock references (here Symmetry Adapted Cluster-Configuration Interaction results). An experimentally well-characterized intermolecular proton transfer reaction has been considered as test case. The computed potential energy profiles were analyzed both in the gas phase and in toluene solution, here represented as a polarizable continuum model. The presence of intermolecular (dark) and intramolecular (bright) charge transfer excited states, whose polarity strongly differs along the reaction pathway, makes clear that only subtle compensation between spurious electronic effects—related to the incorrect asymptotic behavior of the functional—and solvent stabilization of polar states leads to the overall correct description of this excited state reaction when using global hybrids with low percentage of Hartree Fock exchange.