Quantifying partial hole-particle distance at the excited state: A revised version of the DCT indexChemical Physics Letters 2019 DOI: 10.1016/j.cplett.2018.10.060 A modified version of the DCT index, based on total density analysis and...
Implicit solvation using a generalized finite-difference approach in CRYSTAL: implementation and resJournal of Chemical Theory and Computation 2018 DOI: 10.1021/acs.jctc.8b00762 We present the implementation of an implicit solvation...
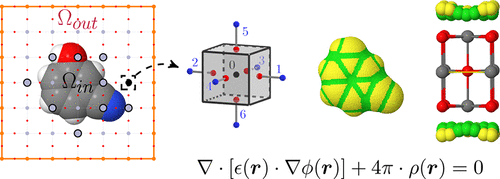
Aggregation-caused quenching versus crystallization induced emission in thiazolo[5,4-b]thieno[3,2-e]Physical Chemistry Chemical Physics 2019 DOI: 10.1039/c8cp04730h We report a QM (TD-DFT) and QM/QM′ (ONIOM) study of the modulation of...
Photophysical Properties of Fluorescent Imaging Biological Probes of Nucleic Acids: SAC-CI and TD-DFJournal of Computational Chemistry 2018 DOI:10.1002/jcc.25553 Recently, exciton‐controlled hybridization‐sensitive fluorescent...




![Aggregation-caused quenching versus crystallization induced emission in thiazolo[5,4-b]thieno[3,2-e]](https://static.wixstatic.com/media/2847f5_556ea8c45c804a91a36d5a07f4221c1a~mv2.jpg/v1/fill/w_249,h_189,fp_0.50_0.50,q_90,enc_auto/2847f5_556ea8c45c804a91a36d5a07f4221c1a~mv2.jpg)

